
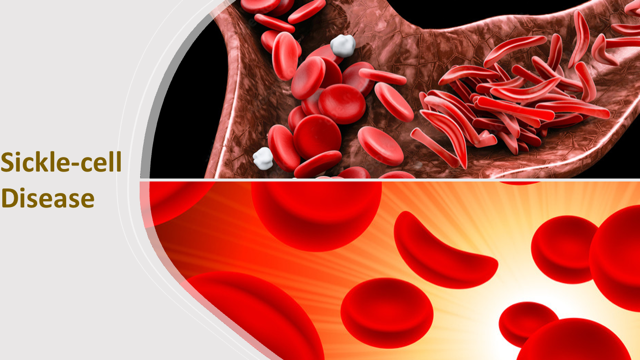
Sickle-cell disease (or SCD) is an inherited, lifelong blood disorder, which affects predominantly Africans, Caribbeans, Middle Easterners, Asians and Hispanics, including those with above ancestry. The disease also occurs in several parts of the Mediterranean regions. Sickle-cell disease is an umbrella of a range of inherited disorders affecting the red blood cells. The most common of these are sickle-cell anaemia (SS), sickle haemoglobin-C disease (SC), sickle beta-plus thalassemia and sickle beta-zero thalassemia. About a hundred million people worldwide are harbouring the sickle-cell traits.
Join our WhatsApp ChannelIn sub-Saharan Africa, more than 200,000 children are born annually with SCD and about 90% of these babies die before their fifth year. The life expectancy of sufferers who survived infanthood and make it into adulthood, averages under 50 years. However, a report in the 2016 edition of the journal, Blood, did mention the possibility of some people with mild sickle-cell conditions living up to 86 years with proper SCD management regime in place, including blood transfusions and strict adherence to medication and hospital appointments. This SCD management regime is augmented with strong family ties.
READ ALSO: New Research Finds Cure For Spinal Cord Injury
The most serious symptom of SCD is anaemia caused by limited oxygen-carrying capacity of the red blood cells. This results in tiredness and shortness of breath. Sickle-cell episodes occur when the blood vessels become blocked by the abnormally shaped red blood cells. The excruciating pain experienced by sufferers can last as long as a week. Other problems attributed to SCD include lung problems, organ damage, strokes, and even delayed growth.
The protein, β-globin (or beta-globin), is a major component of haemoglobin, which red blood cells use in binding and transporting oxygen. In the case of sickle-cell anaemia, a mutation in a single nucleotide of the gene encoding the β-globin (in which the amino acid, glutamic acid or glutamate is replaced by valine), leads to misfolding and assemblage of the protein into fibrous aggregates. The red blood cells become deformed and assumed inflexible crescents, which makes it difficult for the cells to circulate through the blood vessels. The deformed red blood cells (or sickled cells) clog arteries and the walls of blood vessels, and in the process limit oxygen supply to organs. The blockage causes pains that can be likened to that of a heart attack and is known as sickle-cell episode (or vaso-occlusive crises).
Hydroxyurea is a drug used for the treatment of certain cancers and is the only drug approved for treating SCD up until 2017. It was the first drug approved for treating SCD by the US Food and Drug Agency (FDA) in 1998. Hydroxyurea increases the size of red blood cells and causes the cells to stay rounder for longer periods, making it possible for the cell not to sickle easily. But it does cause unpleasant side effects in some people, which include stomach pains, constipation, diarrhoea, skin ulcers, and skin rashes. People harbouring the sickle-cell trait (AS) have partial protection against malaria caused by the parasite, Plasmodium falciparum. However, it is “suspected” that hydroxyurea reduces the protection against malaria conferred by the sickle-cell trait and instead shifts the malaria disease episode from relatively mild form to severity and death. But it has also been reported that hydroxyurea treatment generates nitric oxide (NO), which confers protection against severe malaria in animals and humans. Therefore, the risks and benefits of hydroxyurea vis-à-vis malaria, is unknown.
But currently, there are a slew of drugs in the market for treating SCD including voxelotor, which boosts the oxygen-carrying capacity of haemoglobin, and in some cases improves the shape of the deformed red blood cells. L-glutamine is an antioxidant, which by reducing oxidative stress brings about fewer episodes of sickle-cell-associated pain. The US FDA has approved both these drugs for use against SCD in adults and some children.
The use of gene therapy in the fight against SCD is currently making headways and helping sufferers live longer. A gene therapy called haematopoietic stem cell (HSC) transplantation is revolutionising the treatment of SCD, including HIV/AIDS and other genetic disorders. This technique leverages upon existing techniques like CRISPR-Cas9 gene-editing tool and the use of viruses as vectors for gene delivery.
Haematopoietic stem cell (HSC) gene therapy is based upon the use of bone marrow, which is the ancestry of all blood cell types. This means that the basic premise of success is dependent upon finding healthy donors who are immunologically compatible with recipients. But this donor obstacle is now overcome by the application of gene therapy combining HSC with gene-editing tools or with viral vectors for gene delivery so that patients become their own HSC donors. According to an article in the August 2021 edition of the journal, Nature, patients are “treated with a drug to stimulate the release of haematopoietic stem cells (HSCs) from the marrow into the circulation. The cells are harvested and genetically modified in the laboratory. The person then receives chemotherapy drugs that kill off their remaining natural HSCs. This creates room for the re-implantation of modified stem cells, while culling [destroying] HSCs that might continue to produce sickle-shaped red blood cells. Importantly, this need not be a complete replacement — transplantation studies have shown that an infusion comprising just 20% healthy HSCs is sufficient to reverse the disease.”
As mentioned previously, a mutation occurring in the β-globin protein leads to the fibrously deformed nature of the red blood cells otherwise known as the sickle-cell condition. The effects of the mutation can be reversed in two ways: β-globin gene function can be restored by delivering a new copy of the β-globin gene to another area of the HSC genome or by just correcting the mutated gene. Alternatively, the β-globin gene can be replaced with another protein called γ-globin (or gamma-globin). This particular protein, also known as foetal haemoglobin, does the same work as the β-globin except that it is normally produced only during foetal development. Its work is to assist in ensuring steady oxygenation of blood for the baby in the womb. It does this by transporting oxygen from the bloodstream of the mother to foetal organs and tissues. The gene for γ-globin is permanently turned off in most people few months after birth.
For years, research in SCD was either slowed down or non-existent because it was viewed as an ethnic minority disease. Drug giants were also reticent in tackling the problem for fear of reduced profit margins. But with a slew of technological approaches now in the pipeline, the fight against SCD is gathering momentum. This momentum is helped by a change of policy, especially in the USA, and increase in research fundings.
There are currently 11 treatment centres in Ghana run by Novartis Africa Sickle-Cell Disease Programme. Since 2019 when the programme started, more than 2,000 patients have been treated so far bringing the total treatments with hydroxyurea to little more than 60,000. The programme is aimed at expanding to 9 other African countries by the end of this year. Novartis has already registered hydroxyurea for the treatment of SCD in Uganda, Kenya and Tanzania.
With the multifaceted approach in tackling SCD combined with efficient delivery platforms, positivity has been injected into people with SCD and it is just a matter of time before the disease is brought under control, if not eliminated.


![Relishing Ugba [Oil Bean] Delicacy](https://www.primebusiness.africa/wp-content/uploads/2025/06/Ugba-delicacy-720x480.jpg)









Follow Us