
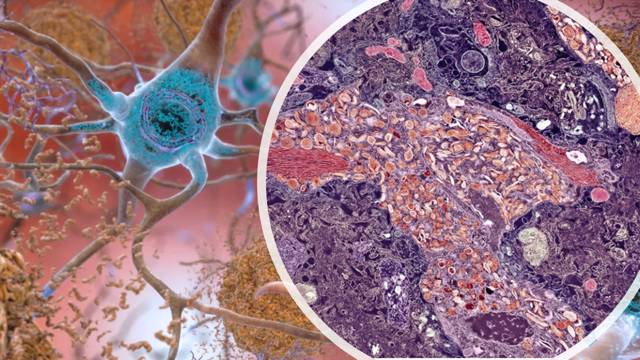
A genetic mutation could be a blessing in disguise, protecting one from the early onset of Alzheimer’s.
An extended family whose members develop Alzheimer’s in their forties or earlier has been the subject of study for Francisco Lopera, a neuroscientist from the University of Antioquia in Medellín, Colombia. Members of the kindred, about 6000 strong, and divided into 25 families carry a genetic variant called the Paisa mutation (or PSEN1 E280A), which invariably leads to early-onset Alzheimer’s disease.
Join our WhatsApp ChannelThe Paisa mutation was named after this extended family group from Antioquia in Colombia, where most carriers of the mutation live. The family group live mostly in an isolated region in the Andes Mountains. There has also been a report of the mutation occurring in a Japanese family.
RED ALSO: Bone Marrow Replacement Therapy May Be A Cure For HIV/AIDS
The Paisa mutation was the most common cause of familial early-onset Alzheimer’s. The carriers of the Paisa mutation have been reported to develop memory loss in their 30s, which is soon followed by cognitive declines, including difficulty in speech by age 45, and full-blown dementia by age 50. Death is usually between 9 and 12 years after the onset of dementia.
Lopera and coworkers writing in the May 2023 edition of the journal, Nature Medicine, reported the identification of a family member of this kindred group who had a second genetic mutation that conferred protection against Alzheimer’s until age 67. The man who had a 4-year formal education and retired at age 60, was only diagnosed with limited verbal learning skills and language difficulties at age 67. His condition progressively deteriorated by age 72 to mild dementia, and to moderate dementia by the time of his death at 74. He died of aspiration pneumonia.
His sister who also had a second genetic mutation as the elder brother, did not have the same level of protection from Alzheimer’s as her brother. She developed cognitive decline at age 58 and was diagnosed with dementia at age 61. She died of sepsis of pulmonary origin at age 73.
Alzheimer’s disease is traditionally believed to be caused by amyloid plaques. Closely associated with the amyloid plaques is a protein called tau. Tau protein forms pathological tangles in the brains of those with Alzheimer’s, accumulating in greater quantities the more the disease progresses.
The autopsy report shows that the man’s brain looked just like the brain of a person with severe dementia except that a little area of the brain called the entorhinal cortex which is responsible for skills coordination such as memory and navigation, showed low levels of the tau protein!
READ ALSO: New Gene-therapy Gel Heals Rare Genetic Skin Wounds
In-depth genetic and molecular analyses revealed a rare and new genetic variant in the Reelin gene, which the workers called RELN-COLBOS (RELN = the Reelin protein; COLBOS = Colombia–Boston Biomarker Research Study where the man was enrolled at age 73 for examination). Reelin is produced in the brain before and after birth, and it functions to regulate brain cell development and function. Even in adulthood, Reelin continues to play important roles in the brain, for example, there are reports linking mutations in Reelin to epilepsy, autism and bipolar disorder. In fact, in animal models, mutations in Reelin leading to a decrease in protein expression do produce features seen in human schizophrenia.
Reelin’s role in Alzheimer’s is not fully understood, therefore the workers transformed the same Reelin mutation into mice. They discovered that mutation in Reelin chemically modified the tau protein, causing it to cluster less around the neurons. In other words, the Reelin-COLBOS variant protected against the accumulation of tau protein, a contributory factor in Alzheimer’s.
This study suggests that amyloid plaques may not be the only cause of Alzheimer’s. And this is not surprising given the fact that many drugs approved for the treatment of Alzheimer’s by the US Food and Drug Administration, are only marginally effective. In other words, there could be many subtypes of Alzheimer’s driven by many factors other than amyloid plaques. This is exemplified by the link to tau protein, which plays a role in mental decline and to which there are clinical trials of therapies targeting the protein.
The hippocampus, which is the region of the brain controlling learning and memory, is smaller in Alzheimer’s patients compared with normal brains. This is due to the degenerative effect of the disease. But despite this condition, the cognitive abilities of the man having this second mutation (ie. RELN-COLBOS) were not affected, suggesting a repurpose of neurones from other parts of the brain to make up for the damage. If indeed this was the case, new therapeutic strategies could be exploited to fight Alzheimer’s, especially in people without the protective effect of the RELN-COLBOS mutation.


![Relishing Ugba [Oil Bean] Delicacy](https://www.primebusiness.africa/wp-content/uploads/2025/06/Ugba-delicacy-720x480.jpg)











Follow Us